CONVUlSIONES
tratamiento
Medicamentos anticonvulsivos
DIAZEPAM
Via
Via
IV,IO
0,2-0,5 mg/kg
MIDAZOLAM
Via
IV,IO
0,15-0,20 mg/kg/dosis
se puede repetir 2 veces
Perfusión: 0,05-0,5
mg/kg/h
IM
0,1-0,3 mg/kg
IR
0,15-0,3 mg/kg/dosis
R
0,5 mg/kg
15-20 mg/kg
se puede repetir otra
dosis de 5-10 mg/kg
Max: 35 mg/kg ó 1 g
FENITOINA
FENOBARBITAL
Via
IV, IO
Niños: 15-20 mg/kg
se puede repetir otra
dosis de 10 mg/kg
Max: 40 mg/kg ó 1 g
IV, IO
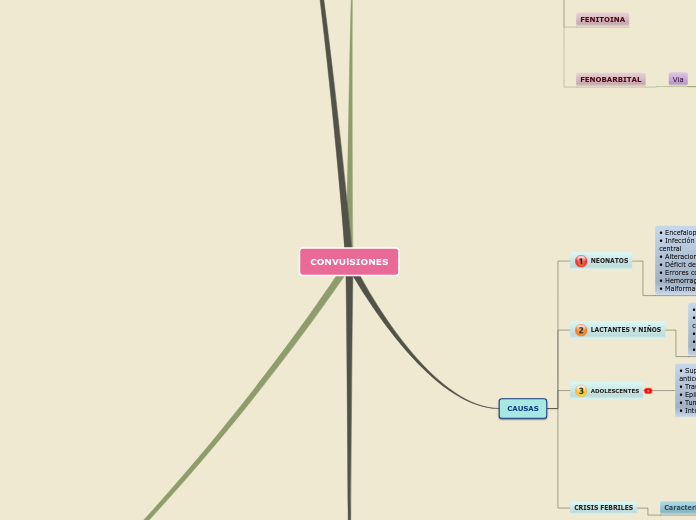
CAUSAS
NEONATOS
• Encefalopatía hipóxico-isquémica
• Infección sistémica o del sistema nervioso
central
• Alteraciones hidroelectrolíticas
• Déficit de piridoxina
• Errores congénitos del metabolismo
• Hemorragia cerebral
• Malformaciones del sistema nervioso centra
LACTANTES Y NIÑOS
• Convulsión febril
• Infección sistémica y del sistema nervioso
central
• Alteraciones hidroelectrolíticas
• Intoxicaciones
• Epilepsia
ADOLESCENTES
• Supresión o niveles sanguíneos bajos de
anticonvulsivantes en niños epilépticos
• Traumatismo craneal
• Epilepsia
• Tumor craneal
• Intoxicaciones (alcohol y drogas)
CRISIS FEBRILES
Caracteristicas
ocurren entre los 6 meses y 5 años de edad en ausencia de infección intracraneal o alteración metabólica y sin
antecedentes de crisis afebriles
clasificacion
Crisis febriles simples
generalizadas, duración < 15
minutos
Crisis febriles complejas
(focales, duración > 15 minutos, recurrentes en el mismo episodio, recuperación lenta
del sensorio, focalidad neurológica residual
Diagnostico
Punción lumbar
deberá realizarse en los menores de 12 meses y en cualquier niño que presente signos de
meningitis o recuperación lenta del sensorio
EEG
Debe realizarse en las crisis complejas repetidas
Tratamiento
diazepam rectal (0,3 mg/kg/día c/12 horas; max: 10 mg dosis y 48 horas de duración
Son una descarga sincrónica excesiva de un grupo neuronal dependiendo
de su localización se manifiesta con síntomas motores, sensitivos, autonómicos o de carácter psíquico,
con o sin pérdida de conciencia.
SINTOMATICAS O SECUNDARIAS
Desencadenadas por un estimulo
Hipoglucemia
Traumatismos
Fiebre
Infeccion del sistema nervioso central
IDEOPATICAS
Sin estumulo
Caracter recurrente
Epilepsia
Crisis parciales (focales)
Crisis parciales simples
Sin afectación del nivel de conciencia
Motoras
somato-sensoriales
Con síntomas autonómicos
Con síntomas psíquicos
Crisi parciales complejas
Afectacion del nivel de conciencia
Crisis parciales que evolucionan a crisis
secundariamente generalizadas
CLASIFICACIÓN
Crisis generalizadas
• Ausencias
• Crisis mioclónicas simples o múltiples
• Crisis clónicas
• Crisis tónicas
• Crisis tónico-clónicas
• Crisis atónicas (astáticas)
Crisis inclasificables
ACTITUD ANTE UNA CONVULSIÓN
Identificar que se trata verdaderamente de
una crisis convulsiva
Preguntar características de la crisis
Tratamiento de la crisis convulsiva
Estabilización de las funciones vitales (ABC)
Determinación de glucemia (tira reactiva). Extraer sangre para laboratorio1 (electrolitos, pH, gases,bicarbonato, urea, creatinina, niveles de anticonvulsivantes).
Si hipoglucemia: S. Glucosado 25% 2 ml/kg. IV.
Administración de medicación anticonvulsiva
neonatos
fenobarbital
15-20 mg/kg IV en 5-10 min
• Min. 0-5: Diazepam2 0,3 mg/kg IV en 2-4 min. (max: 10 mg) ó 0,5 mg/kg rectal.
En los niños menores de 18 meses debe ensayarse una dosis de piridoxina 150 mg/kg IV (50 mg en
recién nacidos).
• Min. 5-10: Repetir la dosis de diazepam
• Min. 10: Fenitoína 15-20 mg/kg IV (max: 1 g) en 10-20 min (monitorización ECG y TA)
• Min. 20: Repetir la dosis de diazepam (riesgo de depresión respiratoria)
• Min. 30: Fenitoína 10 mg/kg IV o fenobarbital 15-20 mg/kg IV.
Consideraciones generales
una convulsión es una urgencia neurológica que hay que intentar que ceda lo antes posible.
Cuanto más prolongada sea la crisis más difícil será su reversibilidad y peor su pronóstico.
status epilepttico
se prolongan durante más de 30 minutos y tiene perdida de conciencia
Anamnesis
conocer la naturaleza de
la crisis
¿Tiene fiebre?
Descartar
meningitis, absceso cerebral
¿Es la primera convulsión o ya ha tenido más
crisis?
¿Ha podido existir algún factor precipitante de
la crisis que no sea la fiebre?
Examen físico
Valoración del estado general
Cuadros graves
Hipertensión intracraneal
Sepsis
Exploración general.
Buscar signos de infeccion focal
Nivel de la fontanela
Medir perímetro cefálico
Exploración neurologica
Atencion a los signos
Infección intracraneal
Focalidad neurológica
Pruebas complementarias
Estudio metabólico
Muestras de laboratorio
Orina
LCR
Punción lumbar
Debe realizarse en todos los niños menores de 12 meses y con sospecha de meningitis
Tomografía axial computarizada (TAC), resonancia magnética (RM).
Niveles sanguíneos de anticonvulsivantes
Determinación de tóxicos en sangre.
Electroencefalograma (EEG).
Niños con una primera convulsión afebril, en las
crisis febriles atípicas y en los niños epilépticos
en los que el patrón o la frecuencia de las crisis
hayan cambiado
Sindrome de West
1960
Triada Sintomatologica
Espasmos Epilepticos
Identificados por el Dr. West en 1841
Hipsarritmia
identificada por Gibs & Gibs en 1952
Ondas y puntas desordenada que varían en duración y localización
Retraso en el desarrollo
psicomotor
Etiología
Sintomático
Las crisis son el resultado de una o mas lesiones estructurales cerebrales identificables
Factores Etiologicos
Prenatales
- Congenitas
- Anomalias Cromosomicas
- Infecciones (Storch)
- Insulto Hipoxico-Isquemico
Perinatales
- Hipoglicemia
- Daño Estructural
Posnatales
- Infecciones
- Hemorragias
- Traumas
- Paro Cardiaco
- Tumores Cerebrales
Criptogénico
También llamado posiblemente sintomatico; se presumen como sintomáticos pero la causa esta oculta
Idiopático
aquellos casos en los cuales no existe una lesión estructural Subyacente ni anomalías neurológicas, existe predisposición genética, y es dependiente de la edad
Fisiopatología
La Fisiopatología del síndrome de west se desconoce; Pero se han postulado diversas teorías:
- Se ha demostrado que coincide con el periodo crítico de formación de las dentritas y la mielinización
- Participación de la corteza cerebral en su origen
Teorías en relación a los espasmos epilépticos:
- Desequilibrio en los neurotransmisores del tallo cerebral
- Son respuestas inespecíficas de un cerebro inmaduro a cualquier daño
- Participación de las estructuras subcorticales
- Anormalidades en la perfusión y metabolismos corticales
Cuando se asocia con síndrome de down hay anormalidades en el receptor de glutamato
Teorías en relación con la Hipsarritmia
- Desequilibrio en los neurotransmisores del tallo cerebral
- Incremento simétrico de la actividad metabólica del núcleo lenticular y del tallo cerebral
- Vía cortico-cortical a través del cuerpo calloso
Manifestaciones clinicas
- Inicia durante el primer año de vida (con mayor frecuencia entre los 3-7 meses)
Los espasmos se caracterizan por la contracción brusca, generalmente bilateral y simétrica de los músculos del cuello, tronco y miembros. Se acompaña de una breve perdida de la conciencia
Se Dividen En
Espasmos de Flexion
Espasmos de Extension
Espasmos Mixtos
Retraso En El Desarrollo Psicomotor
Es mas evidente en pacientes que previamente han tenido un adecuado desarrollo psicomotor puesto que hacen regresión de su propio desarrollo
(El desarrollo psicomotor puede ser normal en el 55% de los casos)
Perdida del seguimiento visual
perdida de la aprehensión voluntaria de los objetos
Aparición de hipotonía
Otros Sintomas
Crisis Focales
Atónicas
Tónicas
Diagnostico diferencial
Trastornos no epilépticos
- Cólicos del lactante
- Mioclonia benigna de la infancia
- postura de opistótonos (espasticidad)
-reflujo gastroesofágico
Trastornos epilépticos
- Epilepsia Mioclónica del lactante
- Encefalopatía mioclónica precoz
- Síndrome de Ohara
Pronostico
- Mortalidad del 5%
- Retardo mental del 90% (déficit motor, trastornos de la conducta y rasgos autistas)
- Desarrollo de otros síndromes convulsivos del 55% - 60%
- Remisión espontánea: el 86% ha sido por infecciones virales
- Empeora el pronóstico cuando es Sindrome de West es sintomático
- La disminución del metabolismo de glucosa bitemporal aumentó el pronóstico desfavorable y rasgos autistas
- Predictores de pronóstico desfavorable: Espasmos asimétricos (desviación lateral de la cabeza o los ojos con la participación de los miembros superiores)
- síndrome de west + síndrome de down mejora el pronostico que los pacientes que solo presentan síndrome de west
Espasmos En Salvas
- desde 1 hasta 60 veces al dia
- al despertar o antes de dormir
- menos frecuentes durante el sueño (se presenta solo en sueño lento nunca durante el sueño paradójico)