Pharmacokinetics Module 4 - ADME
Absorption
the route a drug takes to reach the blood plasma
Drug must cross multiple polar membranes to reach its target site
Each side of a membrane has it's own proteins expression, particularly transport proteins
Dependent on
lipophilicity
partisan coefficient
Kd= [drugs] in lipid / [drugs] in aqueous
Kd= k1/ k2
K1= rate from water to oil
K2= rate from oil to water
this is a ratio, the higher it is the easier it is for the drug to get into the lipid membrane
Say 10:1
will move 10 times faster into lipid than into water
10 times more lipophilic
K1 is 10 times faster than k2
Say 1:10
Will move 10 times faster into water and into lipid
10 times more hydrophilic
k2 is 10 times faster than k1
why does it matter?
Duodenum is aqueous
cell membrane is lipid
If a molecule kd=10, it will move 10 time faster into the cell membrane, but will be slow to leave
Kd=10 is the same as for every 10 molecules in the cell membrane, there is one in the duodenum and one in the blood
Drug should be between these so they do not get trapped in either cell compartment
If a molecule kd=0.14, it will move 14 time faster into the duodenum and the blood, but will not want to cross the membrane
kd=.14 is the same as for every 14 molecules in the duodenum and blood, there is one in the cell membrane
Blood plasma is aqueous
Protein binding
binds to protein in the blood (aqueous)
Reservoir for drug in the blood
can release drug from protein when free drug in blood is used up
Causes a shift in the equilibrium as the concentration for free drug in the blood drops
shift rate to favor moving molecule from the protein into the blood
the level of binding depends on the affinity of the drug for that plasma protein and extent of binding can range from 1 or 2 % up to 99%
most common albumin, but also various lipoproteins, glycoproteins and beta globulins
Increase protein binding increase lipophilicity
Plasma protein binding is saturable
drug saturation of the plasma protein makes a big difference to free drug concentrations
Doubling the dose from 400 to 800 micromolar doesn’t simply double the free concentration of drug in the circulation, it increases it almost exponentially
as less drug reaches the circulation is bound by the plasma proteins. Thus the unbound, free drug is increasingly available for subsequent reaction.
Subtopic
pH and ionization
ionization is the chance of a drug being in its uncharged or charged state
Tendency to Ionize
reversible
Dependent on the pH
Acidic antibiotics, the higher the pH the higher that chance it will ionize
stomach ph=2
acidic antibiotics will not ionize
retains it's uncharged form (acidic), and thus can exit its membrane space
duodenum pH= 6
ionizes the antibiotics (becomes like COO-)
can't cross out
Ion trapping
acidic antiobody accumulates in the duodenum
blood pH= 7
ionizes the antibiotic(becomes like COO-)
acts as a reservoir for drug in the blood
none of the drug can cross back into the membrane (Ion trapping)
drops the amount of drug in the duodenum since it cannot go back (less drug available to active transport)
Drug accumulates in the blood plasma
laws of mass action
used to describe a dissociation constant for these two states at equilibrium
rearranged we can get this equation
rearraged for a third time gives us these equations we can use to find the ratio of ionized to unionized molecules
note that we typically don't need exact percentages of protonation
we can use a protonation table, where pH-pka= HA as a fraction of total drug (or 1/ (10 ^ (pH-pka) +1)
BH+ counts as an ionized drug, thus less likely to be reabsorbed
AKA- protonation of an acid
tends to make the molecule neutral whilst protonation of a base charges that molecule.
This table take HA, the uncharged form, as protonated
Subtopic
pka= known for most drugs and most bodily fluids
ionized drugs tend to dissolve well in aqueous environments, while uncharged molecules can ravel well though lipid environments
aqueous fluids whilst uncharged forms of drugs, will more easily pass through lipid
membranes
Distribution
how the drug is passed along tissues and compartments
compartments
cytoplasm the chief reservoir for water soluble drugs
35%
plasma (5%) , body fat (20%) and interstitial water (15%)
act to a greater or lesser degree as reservoirs for drug dissolution
Trans‐cellular fluid
cerebrospinal, intraocular, peritoneal, pleural and synovial fluids and
digestive secretions
2%
volume
Apparent volume of distribution, apparent because in reality it doesn’t correspond to any real volume in the body.
assumes that there are two places for drug to reside inside the body: in the circulation and in everywhere else
good estimation of how well a drug is able to leave the circulation and penetrate into tissues
a blood sample can estimate whether a drug is primarily in the tissues
or in the circulation
A high VD suggests that the drug has left the circulation
A low VD suggests that the drug is primarily still in the circulation
D= dose, Co= plasma concentration at time zero
The volume of fluid required to contain the full concentration of the drug dose(D), at the same concentration as present in the plasma
second definition for clarification
Blood brain barrier poses problems in drug distribution
Lacks fenestrations between endothelial cells
tight juctions mean that the drug has to pass two lipid membranes (lumenal and basolateral surfaces of the endothelial cells)
excludes large molcules
high lipophilic drugs can pass
diffusion is the primary method for molecular movement
solutes such as amino acids, glucose, amines and purines, are actively transported across the blood brain barrier by carriers
Excretion
Removal of the drug from the system
mostly occurs in the kidneys
most proteins and plasma protiens cannot pass the glomerular
Subtopic
Subtopic
excretion
usually through kidneys
goes through the glomerular membrane (lipid) by glomerular filtration
Protein retained
if drug is bound to protein, it will not be easily transported into the filtrate
this is a problem for warfarin, as it is 98% of the time bound to protein
rate of excretion of Warfarin will be slower than the rate of excretion of another drug with lower affinity for plasma proteins
more free drug available for ultrafiltration or to simply diffuse across the renal tubule
Drugs that are protein bound may still be
readily excreted via the carrier systems
penicillin is about 80% protein
bound, but almost entirely and rapidly excreted by the organic anion transporters
lose small molecules
free drug excreted
loss of the free drug shifts the equilibrum
causes the protein bound drug to be released to fill the loss concentration
over time all the drug will be excreted
can be reabsorbed
affected by the pH of the urine
Acidic urine reabsorbs more acidic antibodies
alkaline urine will cause more acidic free drug to ionize and be excreted
how urine pH can effect tubular secretions
tubercle secretion
tubular secretion involves specific transporter proteins
99% of water exiting the circulation via glomerular filtration, is reabsorbed as the fluid passes along the tubule
drugs are passively reabsorbed along the concentration gradient created by the
concurrent resorption of water
Lipophilic drugs more able to cross membranes and are proportionately reabsorbed more than polar hydrophilic molecules
thus it can be antagonized
competitively inhibit loss of penicillin from the circulation.
excretion of other substrates can be slowed in this way, these include the non steroidal anti‐inflammatories ‐ indomethacin and naproxen‐, but also drugs as diverse as methotrexate, and antiretroviral Zidovudine and the anti‐influenza drug Tamiflu.
Best described by Clearance
is an indicator of efficiency of drug removal
from the plasma
volume of blood cleared of a drug through an organ per unit time (e.g. ml/min)
measure of the efficiency of the organs of elimination, in particular, the kidney,
and may therefore be considered a function of the wellness state of the patient
if it's low, we may have pathology in the excretory organs
Total body clearance is the sum of all the clearances by various mechanisms
number of drugs which are primarily excreted by the kidney
renal clearance=total body clearance
independent of the drug kinetic parameters, such as volume of distribution, bioavailability and drug half life
thus is a constant
Metabolism
Biotransfromation of the drug
Usually to inactivate it
main site for drug metabolism is the liver
Contains two phases
Phase 1
making it more water soluble, and so less able to be re‐absorbed by the renal tubule
Primarily Oxidation reactions and also reduction and hydrolysis.
Oxidation involves the addition of oxygen resulting in hydroxylation, oxidation, dealkylation or deamination, the net result
being to increase water solubility
mediated by cytochrome system associated with the cell's ER
found mostly in the liver
Cytochrome p450 enzymes are iron bearing haem proteins
a large superfamily with 74 gene families
referred to by the designation CYP followed by a number and letter designation
CYP1, CYP2, and CYP3 are the main drug metabolizers
member of these families generally has a
distinct but somewhat overlapping specificity for substrate, sometimes acting on the same substrate but at
different rates, for example
cytochrome p450 cycle adds oxygen or hydroxyl
individual patient response variation due to genetic variation in p450 enzymes
Subtopic
P450 enzyme levels are also readily regulated by external factors
Grapefruit can downregulate CYP3A4 and so inhibit the breakdown of some drugs
not all metabolized in the liver
neuromuscular blocker,succinylcholine, is metabolized by plasma cholinesterases and alcohol
metabolized by both CYP2A6 and
cytoplasmic alcohol dehydrogenase
more for non‐polar drugs than polar drugs, as they must be able to readily cross the cell membrane in order to reach the cytochrome systems
polar drugs are usually just excreted in the urine
Phase 2
altering its structure such that it reduces its intrinsic efficacy and so aids its excretion
Involves conjugation of site chains to drugs through glucuronidation, sulphation,
glutathione addition, glycine or even water conjugation and also methylation
produces a polar molecule that is readily excreted (inactive product)
groups such as glucuronyl, sulphate, methyl and acetyl are attached to the drug
bilirubin and adrenal corticosteroids are also conjugated by glucuronidation, addition of glucuronic acid
creations uridine diphosphate glucuronic acid (UDPGA), a high‐energy phosphate compound
acetaminophen conjugation creates a toxic byproduct called NAPQI
takes place mainly in the liver,
but also lungs and kidneys
first pass effect
first pass effect is the metabolism of drugs prior to reaching the circulation
absorption from small intestine directly to the hepatic portal vein to the liver
EX. Lidocaine --> highly metabolized by liver enzymes and the first pass through the liver removes most of the active drug
thus lidocaine cannot be administered orally
can occur in Gastric mucosa
pro-drugs
metabloites made by metabolizing a drug that are more potent/active than the parent molecule
also more toxic
ex. acetominophen
Subtopic
mechanism
passive diffusion
majority of drug rely on diffusion
nonspecific movement
specific carriers (carries a single species in the direction of its
electrochemical gradient)
also a majority of drugs rely on
some are passive mediators of diffusion down a concentration gradient (facilitated diffusion)
organic cation transporters (OCTs) are a family of cation transporters with specificity for a large number of cationic substrates that are both endogenous and exogenous
mediate the movement of dopamine and choline but includes
some drugs such as vecuronium, quinine and procainamide
OCT2 associated with nephrotoxicity
found in proximal cells in the kidney and notably has cisplatin, the anti‐cancer drug as one of its substrates
transporter is directional and moves cisplatin from the circulation and into the
renal tubule with no way to move cisplatin back out (accumulation and then nephrotoxicity)
cytoplasm of the renal cell. As there is no specific transporter to remove the cisplatin. this
results in accumulation of the drug and ultimately in nephrotoxicity
Some use ATP in active transport
ATP Binding Cassette transporters (ABCs)
Made of p-glycoproteins
are responsible for many cases of drug
resistance in cancer, as they mediate removal of drugs from cells
In high quantity in the intestine, in the renal tubular brush border membranes and in the bile canaliculi
aquaporins
0.4nm in diameter allowing through only the smallest molecules
Too small for most drugs

Subtopic
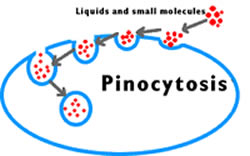
pinocytosis
invagination of a piece of a cell membrane, which forms a vacuole with metabolite within
can release contents in the cell or be moved to the tother side of the cell and released into the extracellular space
only good for large molecules
Subtopic