Application of Next Generation Sequencing (NGS)
Objectives
Background on NGS
Sequencing strategies
Introduce the Linux environment
Background
What is NGS?
a broad term encompasses several modern sequencing technologies
can be largely divided into two based on the length of the sequencing output
short-reads

Illumina-based platform
MiniSeq
MiSeq
NextSeq
HiSeq
NovaSeq

MGIseq
G50
G400
T7

Ion Torrent
Gene Studio S5
Genexus System
long-reads

PacBio
RS II
Sequel
Sequel II
Oxford Nanopore
MinION
GridION
PromethION
A major advantage of NGS compared with PCR is that prior knowledge of the target organism (target-specific primers) is not required.
How is it commonly used in clinical settings?
Risk assessment and screening
e.g. Non-Invasive Prenatal Testing (NIPT)
common trisomy syndromes in fetuses
e.g. Carrier screening for recessive genetic disorders
cystic fibrosis, mendelian inherited disorders
e.g. Germline cancer risk testing
Diagnosis
Diagnosis of specific clinical presentations of suspected genetic diseases
e.g. Neuromuscular disorder
Diagnosis of infectious diseases
e.g. Initial whole-genome sequencing and analysis of the host genetic contribution to COVID-19 severity and susceptibility
e.g. SARS-CoV-2
Prognosis
Therapy selection
What kind of experiments can you do?
Genomics
Whole Genome Sequencing (WGS)
Whole Exome Sequencing (WES)
Targeted Gene
Transcriptomics
Total RNA
mRNA
Targeted
non-coding RNA
Epigenomics
Methylation
ChIPSeq
Many more
Common pipeline
Sequencing strategies
Read depth coverage
What is it and why this is important?
During library prep, the genome is fragmented into short random fragments.
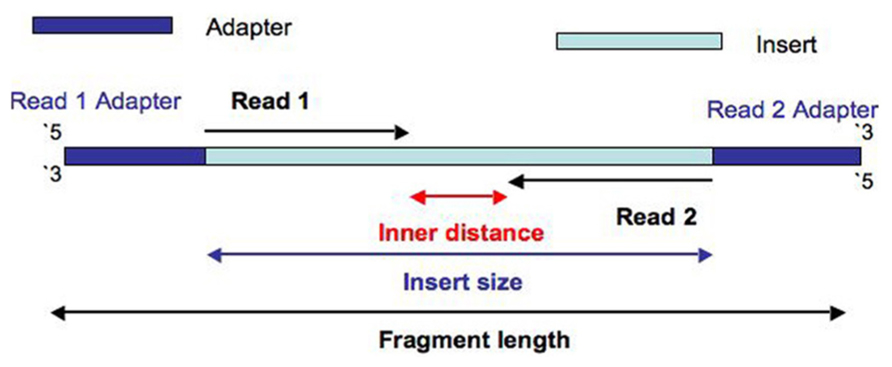
Sequencing adapters are added to these fragments
purpose
Allow sequencer to recognize the fragments
Multiplexing sequencing
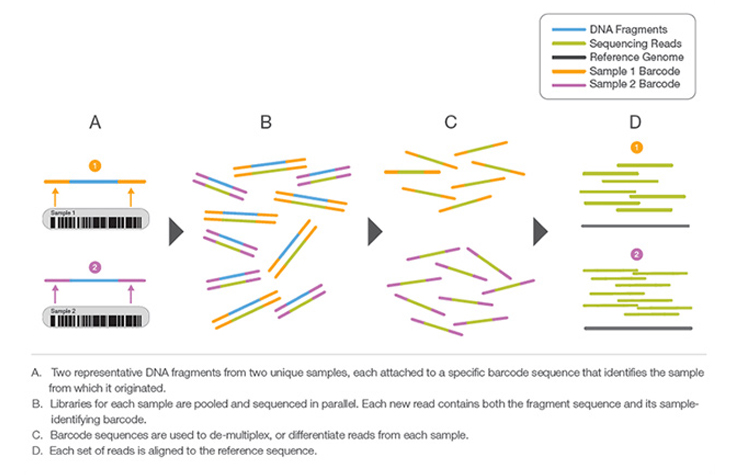
allow sequencing multiple samples in one run
PCR amplify the libraries. ONLY the fragments with the adapters attached are amplified
These random fragments are then sequenced
The reads are then aligned to a reference genome to create longers stretch of sequences
example
To make the tiling process a success
need to have many fragments that overlap between each other
the more overlaps, the higher the alignment confidence
sequencing error will always occurs
result in inability for the tiling process to occur properly
result in erroneous variant calling
but if we have many copies, correct reads will outweigh bad reads
result in high confidence variant calling
e.g. sometimes adapters attach to each other instead to fragment
"low confident" error
"low diversity" error
occurs at the beginning or end of a sequencing read
Depth plays important role for heterogenous samples
e.g. cancer
In a tumor sample, normal cells tend to be observed together with tumor cells
2 populations: Normal and Tumor
We do not know the ratio of each
With high depth, modern bioinformatic tools is able to understand the differences in the reads coverage
fewer reads
might be deletion
more reads
might be duplication
Comprehensiveness
WGS > WES > Gene Panel
Type of variants
SV
WGS LR
SNPs/short InDels
Gene Panel = WES = WGS
Turnaround time
Gene Panel > WES > WGS
Cost
Gene Panel ~ WES > WGS
PRE-ALIGNMENT QC
The most important step
Common questions
Do I have enough sequencing reads?
Depth = (Read length x Number of reads) / Genome size
Did the sequencing work?
During sequencing
e.g. Sequencing Real Time Analysis
Number of reads
platform specific
NextSeq
> 300M
HiSeq
> 100M
Miseq
>25M
Percentage >Q30
in 70% of reads
Error rate
illumina
less than 0.5%
Pacbio
HiFi/Sequel II
less than 1%
Sequel/RSII
less than 15%
Demultiplexing
all libraries should be well-balanced
Post-sequencing level
Reads quality assessment
FastQC
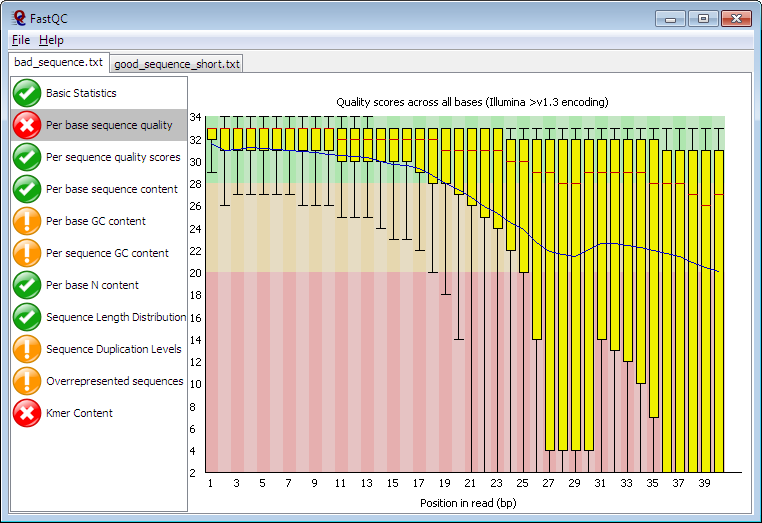
All are important but the most important ones are
Per base sequence quality
check quality of your sequence
Per base GC content
check if there is problem with library
MultiQC
an "advanced" version of FastQC
Can view QC of multiple samples at the same time
can also used as QC post alignment
Sample contamination/swap
contamination
reads contain mixture of DNA from different samples
swap
reads contain DNA from another sample
How could this happen?
many reasons
mislabeling of sample sheets
rotated sample plate
What are the effects?
e.g. in Trio analysis
mistakenly identify variants in VCF as de novo mutations, when the variants actually came from someone else
e.g. in Cancer analysis
calling contaminant germline variants as somatic
tools
verifyBamID
Picard CrosscheckFingerprints
Sample relatedness
check how different samples are related
especially important for Trio-based analysis
verify that the proband dataset is indeed the child of the parental dataset
tools
KING
ALIGNMENT
Koboldt, D.C. Best practices for variant calling in clinical sequencing. Genome Med 12, 91 (2020)
Critical phase
all downstream analyses and interpretation result from the quality of alignment
Raw sequence data are aligned to the reference sequence
e.g. of reference genome
GRCh37/38
CHM13
output is a mapping file called SAM/BAM
which reads mapped where in the ref genome
Tools
BWA-MEM
for short-reads
Minimap2
for long-reads
Alignment QC
Identify PCR duplicates
What is PCR duplicates?
Duplicates resulting from error during PCR
if one small error was introduced during PCR process
This error will be amplified
left with lots of reads that contains these error
tools
Picard MarkDuplicates
Samblaster
additional pipeline
Pathway
once we identified the variants, we want to find out look at the pathway that it is involves in
Free/Open-source
BioCyc
HumanCyc
REACTOME
KEGG
Paid
Ingenuity Pathway Analysis
Pathway Studio
Human Genetics Reference
e.g. identify mode of inheritance (MOI) of the disease/phenotype
e.g. associated genes and known pathogenic variants
Genetics Home Reference
Raredisease.gov
OMIM
Other resources
Disease specific DB
Cancer
TCGA
COSMIC
co-localization GWAS and eQTL
QTLtools
COLOC
eQTL/sQTL database
GTEx
eQTL
locus affecting expression
sQTL
variant affecting splicing
eQTL catalogue
other DB
gnomAD
combine all publicly available WGS and WES
Good to see AF for a SNP or SV
dbSNP
all known SNPs as reported by GRCh, NCBI, HapMap and 1000 Genome Project
Good for seeing AF for SNPs
Gene Set Experiment Analysis
given a set of genes, expression data and list of phenotypes
identify statistically significant, concordant differences between two phenotypic states
Enrichment Map
Cytoscape
ClusterProfiler
Functional annotation
Rank
SIFT
POLYPHEN
CADD
DANN
Annotate & Rank
Annovar
Exomiser
PVP
Random Forest Classifier
AnnotSV
for SVs
Annotate
Variant Effect Predictor
SNPeffect
effect on protein structure & interactions
LS-SNP/PDB
Missense3D
SuSpect
SAAPdap
6,216 samples
69,830,209 SNPs
characteristics
throughput
very high, 25-100Gb
error rate
error rate <=1%
reads length
shorter, 100bp - 250bp
cost
cheaper than long-reads
characteristics
reads length
very long
Pacbio 10kb -15kb
ONT 5kb-10kb
error rate
Pacbio
1-10%
ONT
5-15%
cost
Pacbio
~$3000 per human sample
ONT
~$1800-2000 per human sample
throughput
low to medium
Pacbio
5-10Gb
ONT
~5Gb
How it works?
Sample Prep
During sample prep, the genome is fragmented into short random fragments.
Then sequencing adapters are added to these fragments
What is the purpose of these adapters?
Allow the fragments to bind to the nucleotide bases on the flowcell
Multiplexing sequencing
allow sequencing multiple samples in one run
save $$$
Cluster Generation
the fragments hybridize to the surface of the flow cell
DNA polymerase will bind to the hybridized fragment and create a complimentary strand
The original template/fragment is washed away
the newly created fragment then hybridize with the neighbouring oligo nucleotide bases attached to the flow cell
remember there are 2 types of oligo nucleotides on the flow cells
each is complementary to the starting adapters and the end adapters
this amplification process will be repeated
ONLY the fragments with the adapters attached are amplified
Sequencing
The fragments are then sequenced using fluorescent tagged nucleotide
what is Fluorescent probes?
molecules that absorb light of a specific wavelength and emit light of a different wavelength
How Pacbio Sequencing works?
How Oxford Nanopore Sequencing works?
Looking from the above
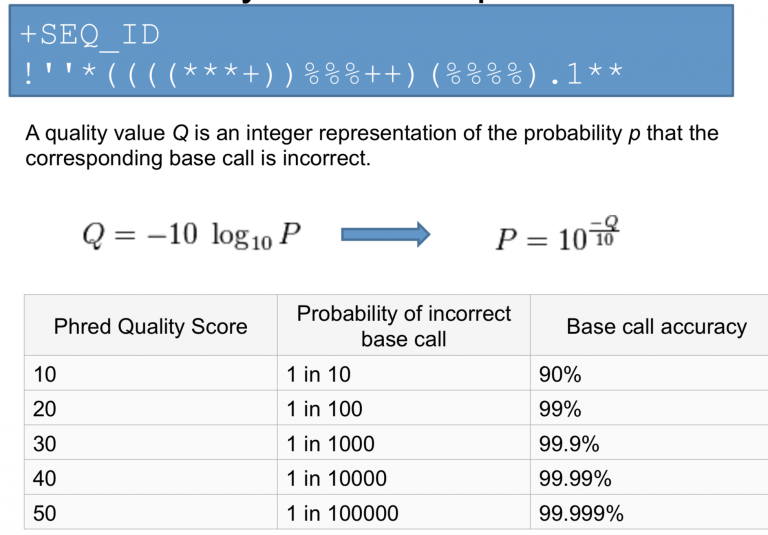
the probability that the intensity represent the incorrect base is stored as Phred score
e.g. if Phred assigns a quality score of 30 to a base, the chances that this base is called incorrectly are 1 in 1000.