FARMACOLOGÍA 1

FARMACODINAMÍA
GENERALIDADES
Describe las acciones de un fármaco en el cuerpo
El complejo fármaco-receptor inicia alteraciones en la actividad bioquímica o molecular de una célula mediante un proceso conocido como transducción de señal
TRANSDUCCIÓN DE SEÑAL
Los fármacos actúan como señales y los receptores actúan como detectores de señales.
El complejo fármaco-receptor
Las células tienen muchos tipos diferentes de receptores, cada uno de los cuales es específico para un agonista particular y produce una respuesta única.
Las células cardiacas también contienen receptores muscarínicos que se unen y responden a acetilcolina.
Este concepto es básicamente similar a la formación de complejos entre enzima y sustrato y comparte muchas características comunes, como especificidad del receptor para un agonista determinado
Estados de receptores
La unión de agonistas hace que el equilibrio cambie de R a R* para producir un efecto biológico.
La magnitud del efecto biológico está directamente relacionada con la fracción de R*.
Principales familias de receptores
Un receptor se define como cualquier molécula biológica a la que se une el fármaco y produce una respuesta medible.
Estos receptores pueden dividirse en cuatro familias:
1) canales iónicos con compuerta de ligandos
2) receptores acoplados a proteína G
3) receptores ligados a enzimas
4) receptores intracelulares
Canales iónicos transmembrana con compuerta de ligando
La porción extracelular de los canales iónicos con compuerta de ligandos contiene el sitio de unión al fármaco.
Dependiendo del ion que se conduce a través de estos canales, estos receptores median diversas funciones, lo que incluye neurotransmisión y contracción muscular
Los sitios de unión a fármacos también se encuentran en muchos canales iónicos con compuerta de voltaje donde pueden regular la función del canal
Por ejemplo, los anestésicos locales se unen al canal de sodio con compuerta de voltaje, lo que inhibe la entrada de sodio y disminuye la conducción neuronal.
Receptores transmembrana acoplados a proteína G
La porción extracelular de este receptor contiene el sitio de unión a ligandos y la porción intracelular interactúa (cuando se activa) con una proteína G
Hay muchos tipos de proteínas G (p. ej., Gs, Gi, y Gq), pero todos los tipos están compuestos de tres subunidades proteínicas.
Un efector común, activado por Gs e inhibido por Gi, es la adenililciclasa, que produce el segundo mensajero adenosina monofosfato cíclico
Receptores ligados a enzimas
esta familia de receptores sufre cambios conformacionales cuando es activada por un ligando, lo que resulta en una mayor actividad enzimática intracelular
Receptores intracelulares:
La cuarta familia de receptores difiere considerablemente de las otras tres en que el receptor es completamente intracelular
Los objetivos primarios de los receptores intracelulares activados son factores de transcripción en el núcleo de la célula que regulan la expresión génica.
Características de la transducción de señal
1) la capacidad de amplificar señales pequeñas
2) mecanismos para proteger a la célula de una estimulación excesiva.
Amplificación de señal
Capacidad de amplificar la intensidad de señal y la duración mediante el efecto de cascada de señal
La unión de salbutamol, por ejemplo, solo puede existir por unos cuantos milisegundos, pero las proteínas G activadas subsecuentes pueden durar por cientos de milisegundos.
existe una pequeña reserva funcional en el corazón con insuficiencia, debido a que la mayoría de los receptores deben estar ocupados para obtener la máxima contractilidad.
Desensibilización y regulación negativa de los receptores
El receptor puede desensibilizarse debido a demasiada estimulación agonista
Durante esta fase de recuperación, se dice que los receptores que no responden son “refractarios”.
La regulación al alta de los receptores puede hacer que las células sean más sensibles a los agonistas o más resistentes a los efectos del antagonista.
Un fármaco se denomina “agonista” si se une a un sitio en una proteína receptora y lo activa para iniciar una serie de reacciones
RELACIONES DOSIS-RESPUESTA
La magnitud del efecto del fármaco depende de la sensibilidad del receptor al fármaco y de la concentración del fármaco en el sitio receptor.
Relación dosis graduada-respuesta
A medida que aumenta la concentración de un fármaco, su efecto farmacológico también aumenta de forma gradual hasta que todos los receptores están ocupados.
Potencia
la potencia es una medida de la cantidad del fármaco necesaria para producir un efecto.
Las preparaciones terapéuticas de los fármacos reflejan su potencia.
Debido a que el rango de concentraciones farmacológicas que causan de 1 a 99% de la respuesta máxima suele extenderse por diversas órdenes de magnitud.
Eficacia
La eficacia es la magnitud de respuesta que causa un fármaco cuando interactúa con un receptor.
La respuesta máxima difiere entre los agonistas totales y los parciales, incluso cuando el fármaco ocupa 100% de los receptores.
Efecto de la concentración del fármaco sobre la unión a receptores.
Fármaco + Receptor ⇆ Fármaco – complejo receptor → Efecto biológico.
Relación de la unión del fármaco con el efecto farmacológico.
1) La magnitud de la respuesta es proporcional a la cantidad de receptores ocupados por el fármaco.
2) el E máx ocurre cuando se unen todos los receptores
3) una molécula del fármaco se une a solo una molécula del receptor.
ACTIVIDAD INTRÍNSECA
Un agonista se une a un receptor y produce una respuesta biológica con base en la concentración del agonista, su afinidad por el receptor y, por lo tanto, la fracción de receptores ocupados.
Agonistas totales
Si un fármaco se une al receptor y produce una respuesta biológica máxima que simula la respuesta al ligando endógeno, se trata de un agonista total
Los agonistas totales se unen a un receptor, estabilizan el receptor en su estado activo y se dice que tienen una actividad intrínseca de uno.
Agonistas parciales
A pesar de ello, los agonistas parciales pueden tener una afinidad que es mayor que, menor que o equivalente a la de un agonista total.
A medida que aumenta el número de receptores ocupados por el agonista parcial.
Las vías dopaminérgicas hiperactivas tienden a estar inhibidas por aripiprazol.
Esto puede explicar la capacidad de aripiprazol para mejorar los síntomas de esquizofrenia, con un pequeño riesgo de causar efectos adversos extrapiramidales
Agonistas inversos
Los receptores no unidos son inactivos y requieren de la interacción con un agonista para asumir una conformación activa.
Esto disminuye el número de receptores activados por debajo de lo observado en ausencia del fármaco .
Antagonista
Los antagonistas se unen a un receptor con una alta afinidad pero poseen cero actividad intrínseca.
Antagonistas competitivos
Antagonistas competitivos:
si el antagonista se une al mismo sitio en el receptor que el agonista en una forma reversible, es “competitivo”.
Antagonistas irreversibles
Los antagonistas irreversibles se unen de forma covalente al sitio activo del receptor, con lo que reducen de forma permanente el número de receptores disponibles al agonista.
Antagonistas alostéricos
Un antagonista alostérico se une a un sitio (sitio alostérico) distinto al sitio de unión agonista y previene la activación del receptor por el agonista.
Antagonismo funcional
Un antagonista puede actuar en un receptor completamente separado, iniciando efectos que son funcionalmente opuestos a los del agonista.
RELACIÓN DOSIS-RESPUESTA CUANTAL
Estas respuestas se conocen como respuestas cuantales, debido a que, para cualquier individuo, ocurre ya sea el efecto deseado o no ocurre.
Una respuesta positiva se define como la caída de al menos 5 mm Hg en la presión arterial diastólica.
Índice terapéutico
El IT es una medida de la seguridad del fármaco, debido a que un valor más grande indica un amplio margen entre las dosis que son efectivas y aquellas que son tóxicas.
Utilidad clínica del índice terapéutico
El índice terapéutico de un fármaco se determina usando estudios del fármaco y experiencia clínica acumulada.
En estos casos, el riesgo de experimentar efectos adversos no es tan grande como el riesgo de dejar la enfermedad sin tratar.
Warfarina
Una mayor fracción de los pacientes responde (para este fármaco, la dosis deseada es un aumento de dos a tres veces en la razón normalizada internacional [RNI]) hasta que, eventualmente, todos los pacientes responden
Penicilina
Para fármacos como penicilina es seguro y frecuente administrar dosis en exceso a lo que se requiere como mínimo para lograr una respuesta deseada sin el riesgo de efectos adversos.
BIBLIOGRAFÍA
1.Karen Whalen,PharmD, BCPS, FAPhA.FARMACOLOGÍA. Pharmacology, Seventh ed.Editores. Rajan Radhakrishnan, Carinda Feild.
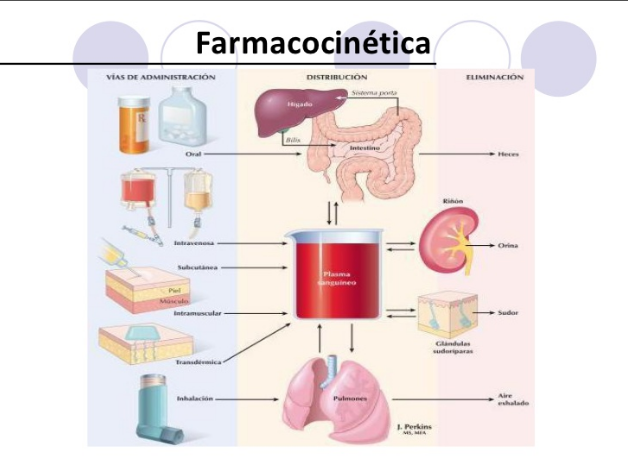
FARMACOCINETICA
GENERALIDADES
La farmacocinética
Se refiere a lo que el cuerpo hace con un fármaco.
La farmacodinamia
Describe lo que el fármaco hace con el cuerpo.
Propiedades farmacocinéticas
Absorción
la absorción desde el sitio de administración permite la entrada del fármaco
Distribución
el fármaco puede dejar el torrente sanguíneo de forma reversible y distribuirse en los líquidos intersticiales e intracelulares
Metabolismo
el fármaco puede biotransformarse a través del metabolismo hepático o de otros tejidos.
Eliminación
el fármaco y sus metabolitos son eliminados del cuerpo en la orina, la bilis o las heces.
VÍAS DE ADMINISTRACIÓN DEL FÁRMACO
ENTERAL
oral.-
Es el procedimiento mediante el cual se suministra por vía bucal medicamentos que tengan acción local o general como tabletas, cápsulas, suspensiones, jarabes etc.
Preparaciones con cubierta entérica
es un recubrimiento químico que protege el medicamento del ácido gástrico
La cubierta entérica es útil para ciertos fármacos (p. ej., omeprazol), que son ácido-lábiles y para fármacos que son irritantes para el estómago, como la aspirina.
Preparaciones de liberación extendida:
tienen recubrimientos o ingredientes especiales que controlan la liberación del fármaco, lo que permite una absorción más lenta y una duración más prolongada de la acción
Por ejemplo, la vida media de la morfina oral es de 2 a 4 horas (h) y debe administrarse seis veces al día para proporcionar un alivio continuo del dolor
Sin embargo, solo se requieren dos dosis cuando se usan tabletas de liberación extendida.
Sublingual/bucal:
implica la colocación del medicamento debajo de la lengua. La vía bucal implica colocar el medicamento entre el carrillo y la encía.
PARENTERAL
Introduce los fármacos directamente en la circulación sistémica.
También se usa para pacientes que no pueden tomar medicamentos por vía oral.
También se usa para pacientes que no pueden tomar medicamentos por vía oral.
PRINCIPALES VÍAS PARENTERALES
Intravenosa (IV)
La administración IV permite un efecto rápido y un grado máximo de control sobre la cantidad de fármaco administrada
Es útil para fármacos que no se absorben por vía oral, como el bloqueador neuromuscular rocuronio
Intramuscular (IM)
pueden estar en soluciones acuosas, que se absorben con rapidez, o en preparaciones de depósito especializadas, que se absorben lentamente.
El fármaco después se disuelve lentamente, proporcionando una dosis sostenida a lo largo de un intervalo extendido.
Subcutánea (SC)
La inyección SC minimiza los riesgos de hemólisis o trombosis relacionados con la inyección IV y puede proporcionar efectos constantes, lentos y sostenidos.
Intradérmica (ID)
consiste en la inyección a la dermis, la capa más vascular de piel debajo de la epidermis
Inhalación oral y preparaciones nasales
permiten suministrar con rapidez un fármaco a lo largo de una gran área de superficie de membranas mucosas de las vías respiratorias y el epitelio pulmonar
Intratecal/intraventricular:
Cuando se requiere de efectos locales y rápidos, es necesario introducir los fármacos directamente en el líquido cefalorraquíde
Tópica:
la aplicación tópica se usa cuando se busca un efecto local del fármaco.
Transdérmica
esta vía de administración obtiene efectos sistémicos mediante la aplicación de medicamentos a la piel, por lo general mediante un parche transdérmico
Rectal:
La vía rectal tiene la ventaja adicional de prevenir la destrucción del fármaco en el ambiente gastrointestinal.
Esta vía también es útil si el fármaco induce el vómito cuando se administra por vía oral o si el paciente está inconsciente.
DISTRIBUCIÓN FARMACOLÓGICA
DEPURACIÓN FARMACOLÓGICA A TRAVÉS DEL METABOLISMO
Vías principales de eliminación
El metabolismo hepatico
La eliminación biliar
La excreción urinaria
Cinética del metabolismo
Cinética de primer orden
La velocidad del metabolismo del fármaco y de eliminación es directamente proporcional a la concentración del fármaco libre y se observa una cinética de primer orden
Cinética de orden cero
Con unos cuantos fármacos, como la aspirina, etanol y fenitoína, las dosis son muy grandes.
Reacciones del metabolismo del fármaco
Fase I
Convierten fármacos lipofílicos en moléculas más polares al introducir o desenmascarar un grupo funcional polar, como –OH o –NH2.
Reacciones de fase I que utilizan el sistema
Nomenclatura
El nombre de la familia está indicado por el número arábigo que sigue a CYP y la letra mayúscula designa la subfamilia, por ejemplo, CYP3A
Especificidad
Debido a que hay muchos genes diferentes que codifican múltiples enzimas, hay muchas isoformas diferentes de P450.
Variabilidad genética
Las variaciones de la actividad P450 pueden alterar la eficacia del fármaco y el riesgo de eventos adversos.
Inductores CYP
Fenobarbital
Rifampicina
Carbamazepina
Inhibidores CYP
Warfarina
Ketoconazol
Omeprazol
Estos incluyen la oxidación de aminas (p. ej., oxidación de catecolaminas o histamina), deshidrogenación de alcohol (p. ej., oxidación de etanol), esterasas (p. ej., metabolismo de la aspirina en el hígado) e hidrólisis (p. ej., procaína).
Fase II
Esta fase consiste de reacciones de conjugación. Si el metabolito de la fase I es suficientemente polar, puede ser excretado por los riñones.
Es el proceso mediante el cual un fármaco deja de forma reversible el torrente sanguíneo y entra al líquido extracelular y a los tejidos.
Flujo de sangre
El flujo sanguíneo elevado, junto con la alta lipofilicidad de propofol, permite una distribución rápida al sistema nervioso central y produce anestesia.
Permeabilidad capilar
Unión de fármacos a las proteínas plasmáticas y los tejidos
Unión a proteínas plasmáticas
La unión reversible a proteínas plasmáticas aísla los fármacos en una forma no difundible y hace más lenta la transferencia fuera del compartimiento vascula
Unión a las proteínas tisulares:
Muchos fármacos se acumulan en los tejidos, llevando a mayores concentraciones en tejidos que en líquido intersticial y sangre
Lipofilicidad
El principal factor que influye sobre la distribución de los fármacos lipofílicos es el flujo de sangre al área.
Volumen de distribución
El volumen aparente de distribución, Vd, se define como el volumen de líquido que se requiere para contener la totalidad del fármaco en el cuerpo a la misma concentración medida en el plasma.
Distribución en los compartimientos de agua en el cuerpo
Compartimiento plasmático
Líquido extracelular
Agua corporal total
Determinación del Vd
Efecto del Vd en la vida media del fármaco
Tiene una importante influencia en la vida media de un fármaco porque la eliminación del fármaco depende de la cantidad de medicamento que llega al hígado o al riñón
DISEÑO Y OPTIMIZACIÓN DEL ESQUEMA DE DOSIFICACIÓN
La selección de un esquema depende de varios factores del paciente y del fármaco, lo que incluye la rapidez con la que tienen que obtenerse las concentraciones terapéuticas de un fármaco.
Esquemas de infusión continua
Concentración plasmática de un fármaco después de infusión IV continua
la concentración plasmática del fármaco se eleva hasta que se alcanza un estado estable
Influencia de la velocidad de infusión sobre la concentración en estado estable
La concentración plasmática en estado estable (Css) es directamente proporcional a la velocidad de infusión.
Tiempo hasta alcanzar la concentración farmacológica en estado estable.
La concentración de un fármaco aumenta desde cero al inicio de la infusión hasta su nivel final en estado estable
Optimización de la dosis
Dosis de mantenimiento
Los fármacos por lo general se administran para mantener una Css dentro de la ventana terapéutica.
Dosis de carga
En ocasiones es necesario obtener con rapidez las concentraciones plasmáticas deseadas (p. ej., en infecciones graves o arritmias).
Ajuste de la dosis
La cantidad de un fármaco administrada para un trastorno determinado se basa en un “paciente promedio”.
Esquemas de dosis fija/tiempo fijo
Las dosis fijas de medicamentos IV u orales administrados a intervalos fijos resultan en fluctuaciones dependientes del tiempo en la concentración circulante del fármaco
Inyecciones IV múltiples
Cuando un fármaco se administra de forma repetida a intervalos regulares, la concentración plasmática aumenta hasta que se alcanza un estado estable
Efecto de la frecuencia de dosificación
Con la administración repetida a intervalos regulares, la concentración plasmática de un fármaco oscila alrededor de una media.
Administraciones orales múltiples
La mayoría de los fármacos administrados de forma ambulatoria son medicamentos orales que se toman a una dosis específica una, dos o más veces al día.
EXCRECIÓN POR OTRAS VÍAS
La excreción farmacológica también puede ocurrir a través
intestinos

bilis
pulmones

leche materna
Los fármacos que no se absorben después de la administración oral de fármacos que se secretan directamente en el intestino o en la bilis se excretan en las heces.
Depuración corporal total
es la suma de todas las depuraciones de los órganos que metabolizan fármacos y de los que los eliminan.
Situaciones clínicas que resultan en cambios en la vida media del fármaco
Cuando un paciente tiene una anormalidad que altera la vida media de un fármaco, se requiere ajustar la dosis.
1) disminución del flujo sanguíneo renal o hepático, por ejemplo, en el choque cardiógeno, la insuficiencia cardiaca o la hemorragia
2) menor capacidad para extraer el fármaco del plasma, por ejemplo, en la enfermedad renal
3) disminución del metabolismo, por ejemplo, cuando un fármaco concomitante inhibe el metabolismo o en la insuficiencia hepática, como en la cirrosis.

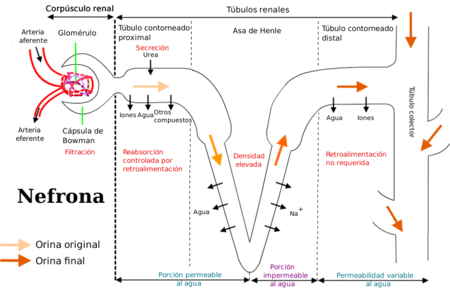
DEPURACIÓN DEL FÁRMACO POR EL RIÑÓN
La eliminación de los fármacos del cuerpo ocurre a través de una variedad de vías; la más importante es la eliminación a través del riñón hacia la orina.
Eliminación renal de un fármaco
Filtración glomerular:
Los fármacos entran al riñón a través de las arterias renales, que se dividen para formar un plexo capilar glomerular.
Las variaciones en la filtración glomerular y la unión proteínica de los fármacos sí afectan este proceso.
Secreción tubular proximal
La secreción ocurre sobre todo en los túbulos proximales por dos sistemas de transporte activo que requieren energía
Uno para aniones (p. ej., formas desprotonadas de ácidos libres)
Uno para cationes (p. ej., formas protonadas de bases débiles).
Reabsorción tubular distal
A medida que un fármaco se mueve hacia el túbulo contorneado distal, su concentración aumenta y excede la del espacio perivascular.
ABSORCIÓN DE FARMACOS
Es la transferencia de un fármaco del sitio de administración al torrente sanguíneo.
Mecanismos de absorción de fármacos a partir de la vía gastrointestinal
Difusión pasiva:
Es el gradiente de concentración a través de una membrana que separa dos compartimientos del cuerpo.
Difusión facilitada
No requiere energía, puede ser saturada y puede verse inhibida por compuestos que compiten por el transportador.
Transporte activo:
Es capaz de mover los fármacos contra un gradiente de concentración de una región de baja concentración farmacológica a una de mayor concentración.
Endocitosis y exocitosis
Este tipo de absorción se usa para transportar fármacos de un tamaño excepcionalmente grande a través de la membrana celular.
Factores que influyen sobre la absorción
Efecto del pH sobre la absorción del fármaco:
la mayoría de los fármacos son ácidos débiles o bases débiles. Los fármacos ácidos (HA) liberan un protón (H+), produciendo un anión (A–) cargado:
Flujo de sangre al sitio de absorción
Los intestinos reciben mucho más flujo sanguíneo que el estómago, por lo que se favorece la absorción intestinal frente a la gástrica.
Área de superficie total disponible para absorción:
con una superficie rica en borde en cepillo que contiene microvellosidades, el intestino tiene un área de superficie de unas mil veces la del estómago, haciendo más eficiente la absorción del fármaco a lo largo del intestino.
Tiempo de contacto en la superficie de absorción
Si un fármaco se mueve a partir del tracto gastrointestinal con gran rapidez, como puede pasar con la diarrea intensa, no se absorbe bien.
Expresión de glucoproteína P:
La glucoproteína P es una proteína transportadora de membrana responsable de transportar varias moléculas, lo que incluye fármacos, a través de membranas
Biodisponibilidad
es la velocidad y grado al cual el fármaco administrado alcanza la circulación sistémica.
Determinación de biodisponibilidad:
se determina al comparar las concentraciones plasmáticas del fármaco después de una vía particular de administración (p. ej., administración oral) con concentraciones que se alcanzan mediante administración IV
Factores que influyen sobre la biodisponibilidad:
Metabolismo hepático de primer paso
cuando se absorbe un fármaco a partir del tracto gastrointestinal, entra a la circulación portal antes de entrar a la circulación sistémica
Solubilidad del fármaco
los fármacos muy hidrofílicos se absorben de forma deficiente debido a la incapacidad para cruzar las membranas celulares ricas en lípidos.
Inestabilidad química:
algunos fármacos, como la penicilina G, son inestables en el pH de los contenidos gástricos.
Naturaleza de la formulación farmacológica:
la absorción del fármaco puede verse alterada por factores que no están relacionados con la química del fármaco.
Bioequivalencia y otros tipos de equivalencia
tienen la misma forma de dosis, contienen el mismo ingrediente activo a la misma potencia y usan la misma vía de administración