Fenilcetonuría
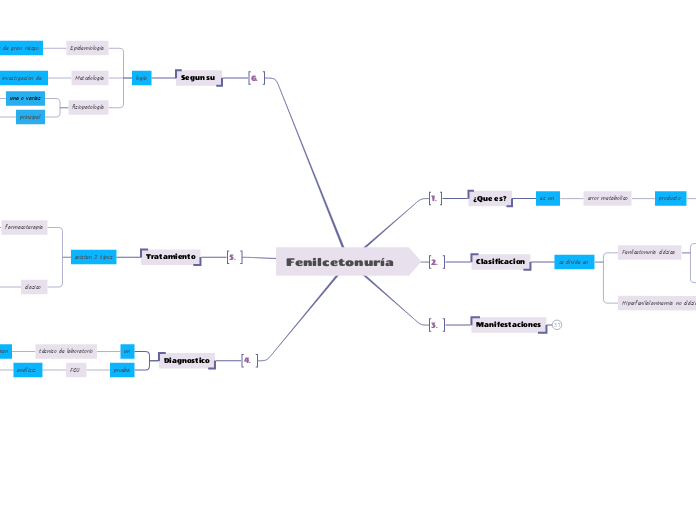
¿Que es?
es un
error metabolico
producto
mutacion gen
encargado
codificacion fenilalanina hidroxilasa
Clasificacion
se divide en
Fenilcetonuria clásica
tiene
Deficiencia completa
de
PAH
concentraciones plasmáticas
de
fenilalanina
en
1200µmol/L
tolerancia dietaría
de
fenilalanina < 350 mg
es
asociada
a
trastorno desarrollo intelectual
si no
se trata
Hiperfenilalaninemia no clásica
grupo de
enfermedades metabólicas
que tiene
niveles elevados
de
fenilalanina en sangre.
Manifestaciones
Segun su
logia
Epidemiologia
factores de gran riesgo
matrimonio entre familiares
antecedentes familiares
Metodologia
investigacion de
fenilalanina hidroxilasa
fin de recopilar
informacion
para darle
importancia al tamizaje neonatal
fisiopatología
una o varias
mutaciones genéticas
determinan
deficiencia o ausencia
de
fenilalanina hidroxilasa
principal
organo afectado
es el
encefalo
por
alteracion de mielinizacion
Tratamiento
existen 2 tipos
farmacoterapia
como
Sapriterina
Aminoácidos neutros largos
PEG-PAL
clasico
como
Restricción dietaría
suplementar con tirosina
mejorar
rendimiento academico
restriccion de proteinas
Diagnostico
un
técnico de laboratorio
extraen
gotas de sangre
del
talón del bebé.
el
laboratorio
analizar
muestra de sangre
para detectar
cantidad de fenilalanina
en muestra
prueba
FCU
análisis
de sangre
recién nacidos
a
3 dias
de
parto